Yalcin B, Nicod J, Bhomra A, Davidson S, Cleak J, Farinelli L, Osteras M, Whitley A, Yuan W, Gan X, Goodson M, Klenerman P, Satpathy A, Mathis D, Benoist C, Adams D, Mott R, Flint J.
PLoS Genetics, 2 September 2010
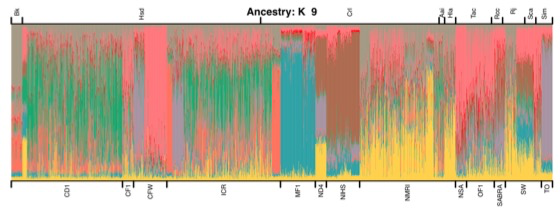
Genome-wide association studies using commercially available outbred mice can detect genes involved in phenotypes of biomedical interest. Useful populations need high-frequency alleles to ensure high power to detect quantitative trait loci (QTLs), low linkage disequilibrium between markers to obtain accurate mapping resolution, and an absence of population structure to prevent false positive associations. We surveyed 66 colonies for inbreeding, genetic diversity, and linkage disequilibrium, and we demonstrate that some have haplotype blocks of less than 100 Kb, enabling gene-level mapping resolution. The same alleles contribute to variation in different colonies, so that when mapping progress stalls in one, another can be used in its stead. Colonies are genetically diverse: 45% of the total genetic variation is attributable to differences between colonies. However, quantitative differences in allele frequencies, rather than the existence of private alleles, are responsible for these population differences. The colonies derive from a limited pool of ancestral haplotypes resembling those found in inbred strains: over 95% of sequence variants segregating in outbred populations are found in inbred strains. Consequently it is possible to impute the sequence of any mouse from a dense SNP map combined with inbred strain sequence data, which opens up the possibility of cataloguing and testing all variants for association, a situation that has so far eluded studies in completely outbred populations. We demonstrate the colonies’ potential by identifying a deletion in the promoter of H2-Ea as the molecular change that strongly contributes to setting the ratio of CD4+ and CD8+ lymphocytes.